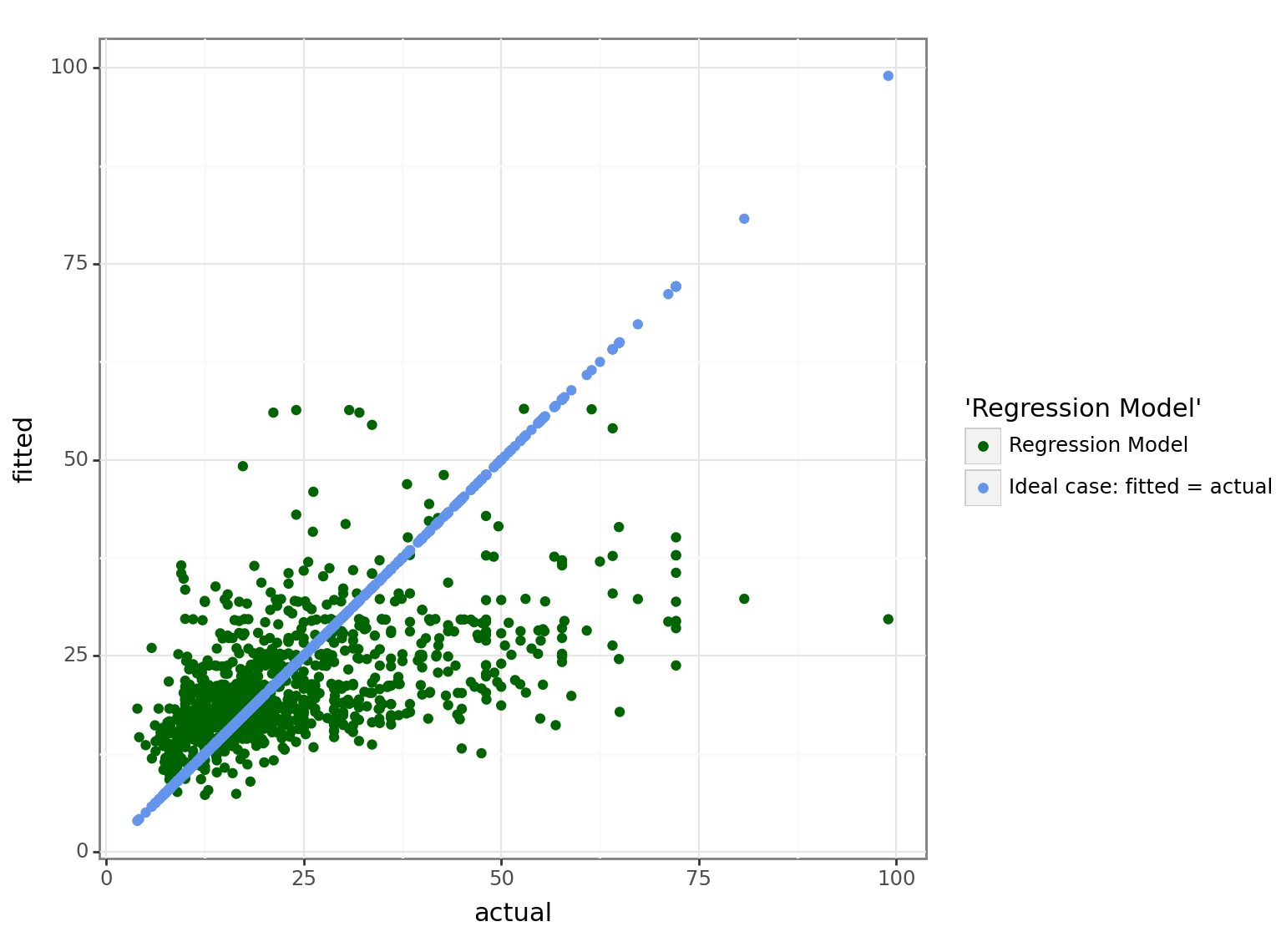
Ideally \(\widehat{Y} = Y\), however, remember that the the conditional expectation is the best predictor, which means that our predictions will have some degree of inaccuracy:
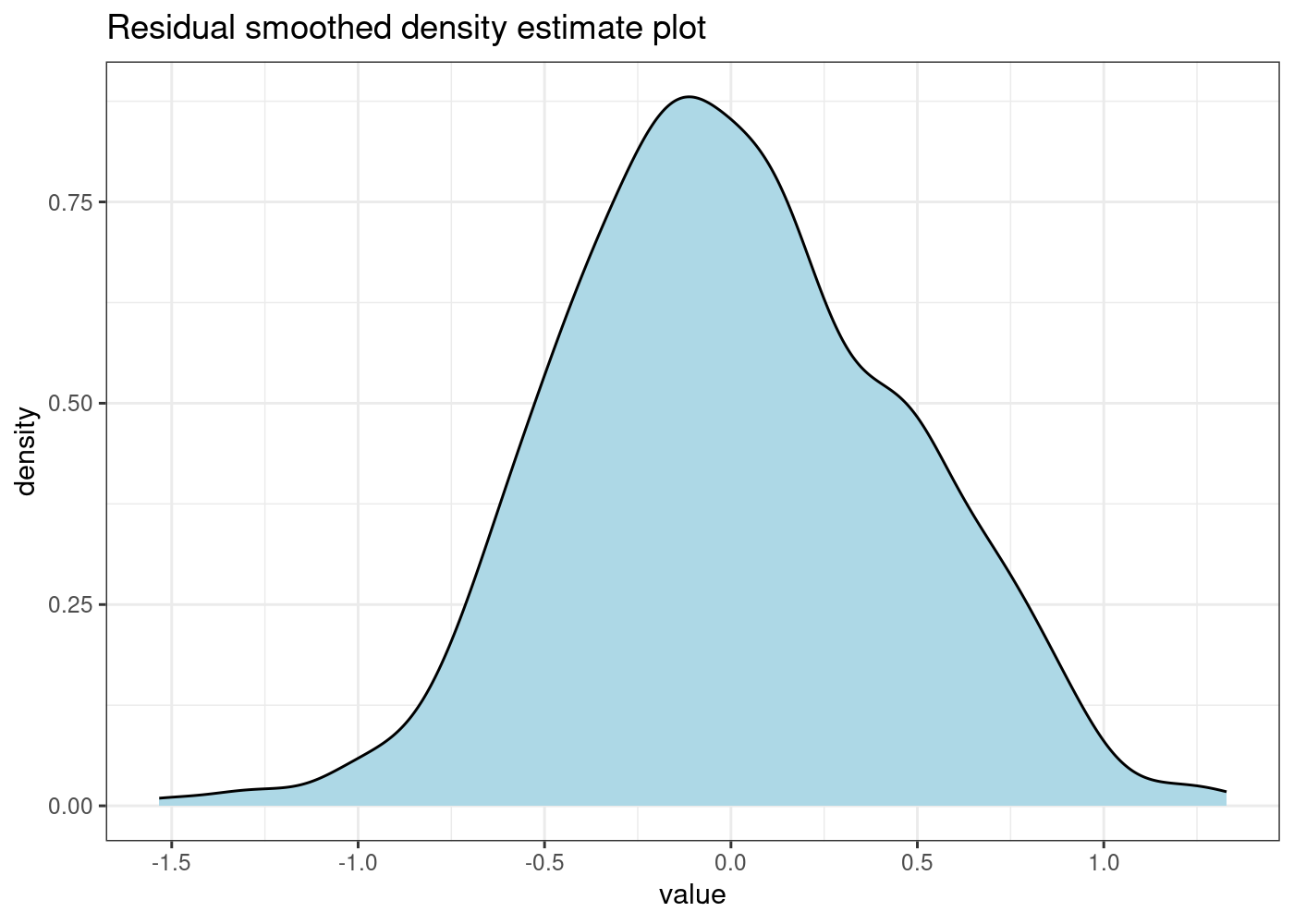
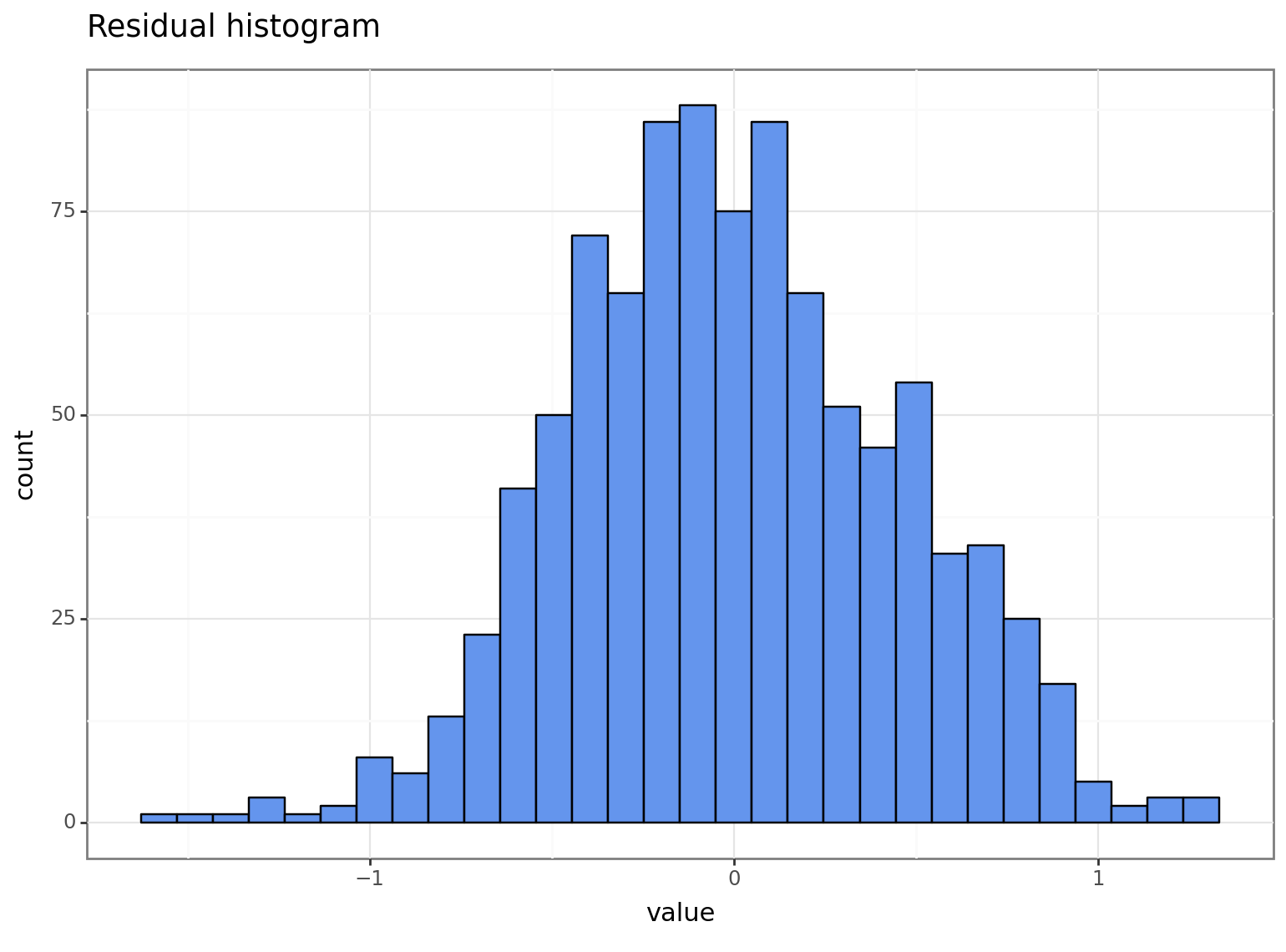
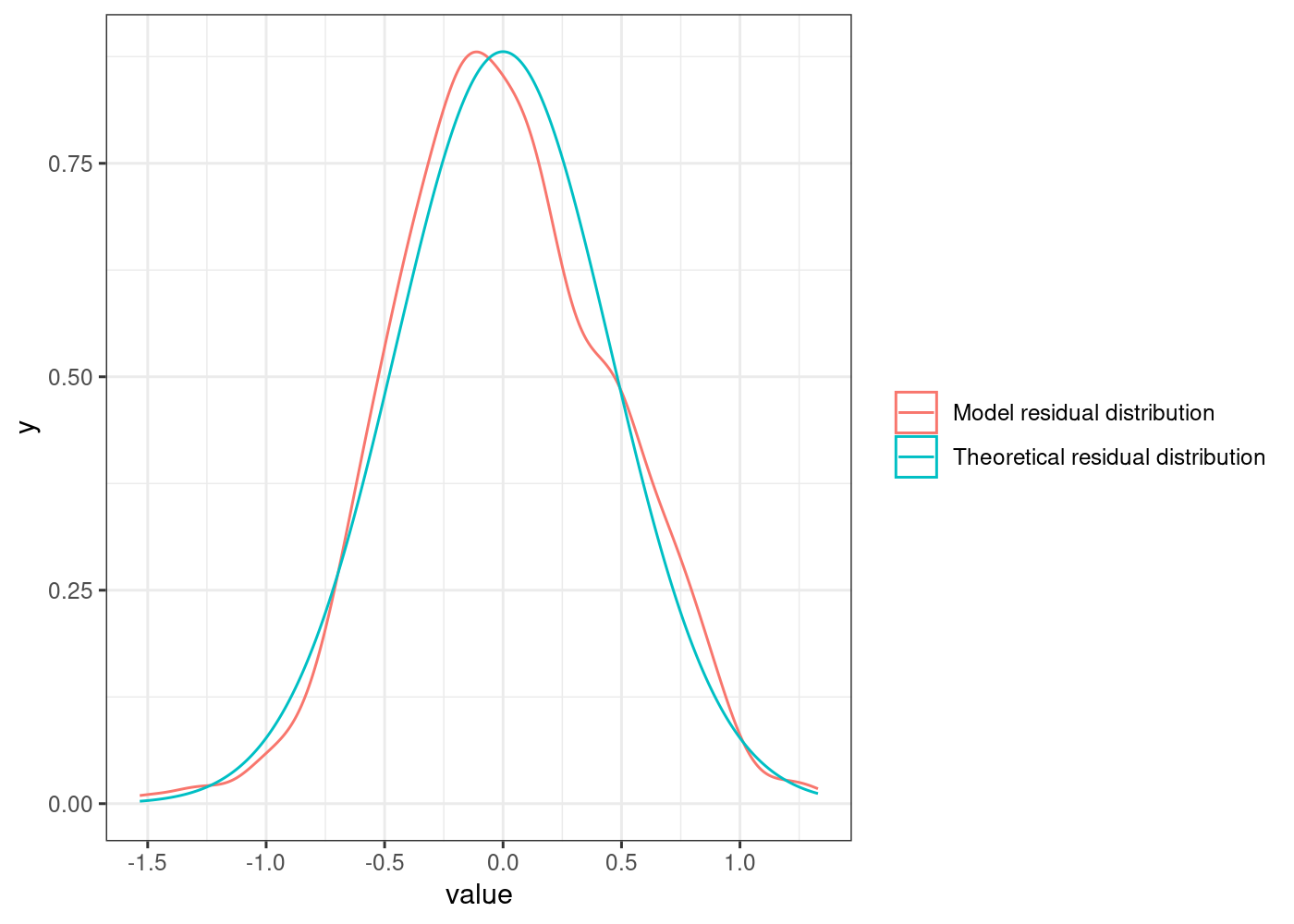
While plotting the “actual vs. fitted” values gives some insight, a more common approach is to analyse the model residuals. The residuals measure the difference between the actual and fitted values. As such, we will begin by visually examining their distribution:
The residuals of our final model appear much closer (at least visually) to a normal distribution.
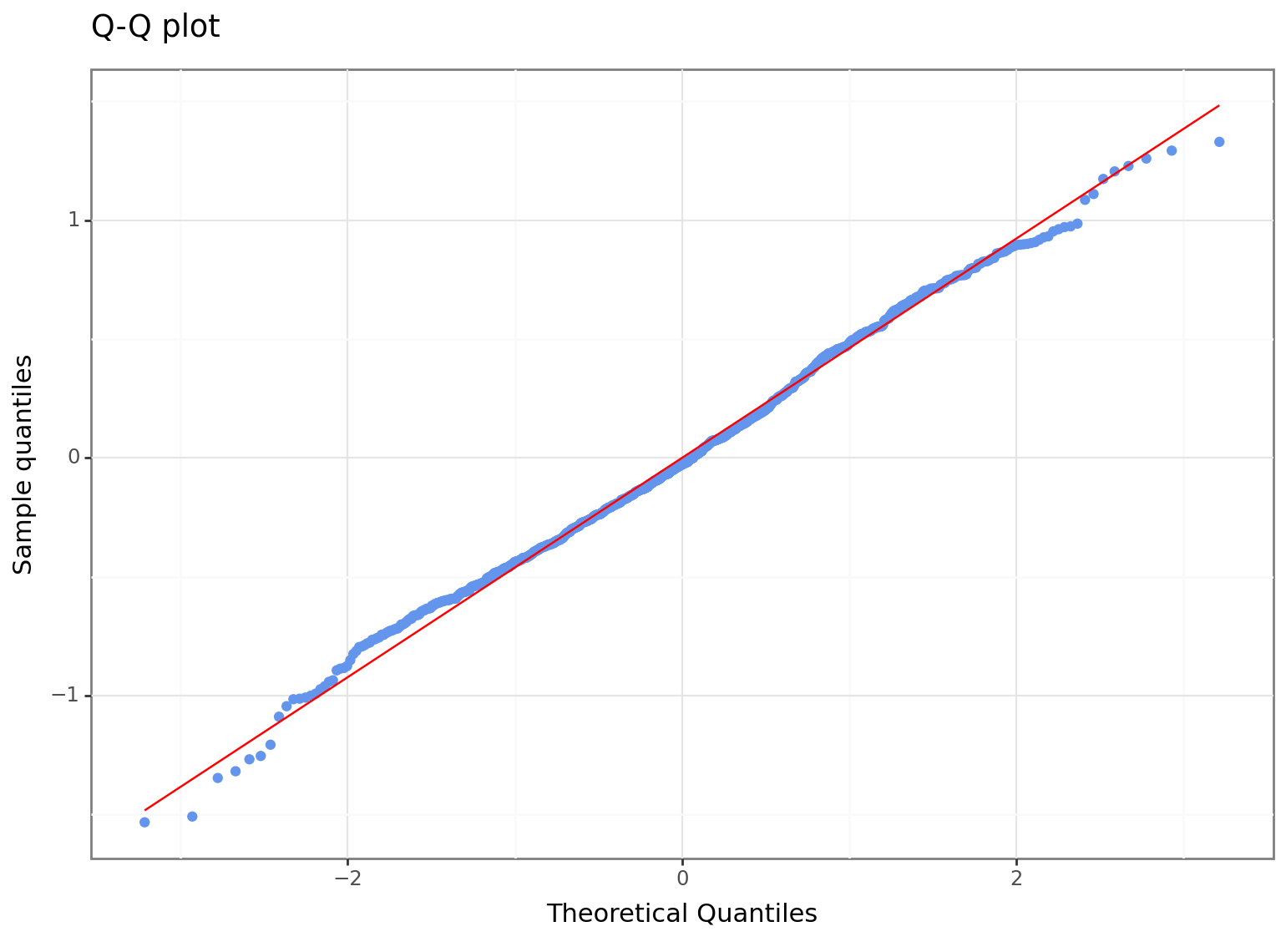
The above plot is only possible since we have a lot of observation to simulate the theoretical distribution. Another way to compare the actual residuals with the theoretical ones is by analysing their quantile-quantile plot:
Here the red diagonal line is for the theoretical quantiles, while the blue dots are the quantiles of the model residuals.
While the graphical analysis gives us some insight, we cannot say for sure whether the residuals follow the theoretical assumptions from the Gauss-Markov theorem. We want to answer the following questions:
How close are our residuals to the normal distribution?
Are the residuals homoskedastic and do they exhibit autocorrelation?
To answer these questions, we will carry our a number of statistical tests.
Residual diagnostics tests
We will check for autocorrelation by testing the following hypothesis: \[
\begin{cases}
H_0&:\text{the errors are serially uncorrelated (i.e. no autocorrelation)}\\
H_1&:\text{the errors are autocorrelated (the exact order of the autocorrelation depends on the test carried out)}
\end{cases}
\]
for(iin1:10){print(paste0("BG Test for autocorrelation order = ", i, "; p-value = ", round(lmtest::bgtest(mdl_4, order =i)$p.value, 2)))}
[1] "BG Test for autocorrelation order = 1; p-value = 0.61"
[1] "BG Test for autocorrelation order = 2; p-value = 0.71"
[1] "BG Test for autocorrelation order = 3; p-value = 0.39"
[1] "BG Test for autocorrelation order = 4; p-value = 0.42"
[1] "BG Test for autocorrelation order = 5; p-value = 0.47"
[1] "BG Test for autocorrelation order = 6; p-value = 0.49"
[1] "BG Test for autocorrelation order = 7; p-value = 0.61"
[1] "BG Test for autocorrelation order = 8; p-value = 0.53"
[1] "BG Test for autocorrelation order = 9; p-value = 0.63"
[1] "BG Test for autocorrelation order = 10; p-value = 0.6"
for i inrange(1, 11): tmp_val = sm.stats.acorr_breusch_godfrey(mdl_4, nlags = i)[1]print(f"BG Test for autocorrelation order = {i}; p-value = {round(tmp_val, 2)}")
BG Test for autocorrelation order = 1; p-value = 0.61
BG Test for autocorrelation order = 2; p-value = 0.71
BG Test for autocorrelation order = 3; p-value = 0.39
BG Test for autocorrelation order = 4; p-value = 0.42
BG Test for autocorrelation order = 5; p-value = 0.47
BG Test for autocorrelation order = 6; p-value = 0.49
BG Test for autocorrelation order = 7; p-value = 0.61
BG Test for autocorrelation order = 8; p-value = 0.53
BG Test for autocorrelation order = 9; p-value = 0.63
BG Test for autocorrelation order = 10; p-value = 0.6
As before - we see that there is no significant autocorrelation, as we have no grounds to reject the null hypothesis in all cases.
Note that carrying out multiple hypothesis tests leads to the multiple comparison problem. In reality, we would not try to re-do the same tests for different autocorrelation orders and instead select an order which would make sense (e.g., maybe the data is ordered by family members, so only \(2\) to \(4\) closest observations could be correlated).
We will now check for heteroskedasticity by carrying out a test for the following hypothesis: \[
\begin{cases}
H_0&: \text{residuals are homoskedastic}\\
H_1&: \text{residuals are heteroskedastic}
\end{cases}
\]
We have that the \(p\)-value \(< 0.05\), so we reject the null hypothesis that the residuals are homoskedastic. Which means that the residuals are heteroskedastic.
Finally, we check the residual normality by testing the following hypothesis:
\[
\begin{cases}
H_0&:\text{residuals follow a normal distribution}\\
H_1&:\text{residuals do not follow a normal distribution}
\end{cases}
\] We will carry out a number of tests and combine their p-values into a single output:
Note that in the above tests there are differences between the results from R and Python due to different implementations of the Kolmogorov-Smirnov and Cramer-von Mises tests. For example, in the scipy.stats.cramervonmises function, we pass the mean and standard deviation parameters, while in sm.stats.diagnostic.kstest_normal the mean and variance parameters are estimated.
Generally, the Shapiro–Wilk test has the best power for a given significance, though it is best to not rely on a single test result.
We see that the \(p\)-value is less than the \(5\%\) significance level for the Anderson-Darling and Shapiro-Wilk tests (and the Cramer-von Mises in R), which would lead us to reject the null hypothesis of normality. On the other hand the \(p\)-value is greater than \(0.05\) for Jarque-Bera and Kolmogorov-Smirnov tests, leading us to not reject the null hypothesis of normality.
Since Shapiro–Wilk test has the best power for a given significance, we will go with its result and reject the null hypothesis of normality.
Overall, our conclusions are as follows:
The model residuals are not serially correlated.
The model residuals are heteroskedastic.
The model residuals are non-normally distributed.
Correcting the standard errors of our model
So, our model residuals violate the homoskedasticity assumption, as well as the normality assumption.
Since our model residuals violate the homoskedasticity assumption - we can correct the standard errors using \(\rm HCE\) (Heteroskedasticity-Consistent standard Errors). This is known as the White correction - it will not change the \(\widehat{\beta}^{(OLS)}\), which are unbiased and ineffective, but it will correct \(\widehat{\mathbb{V}{\rm ar}}\left( \widehat{\boldsymbol{\beta}}_{OLS} \right)\).
While there are many different \(\rm HCE\) specifications, evaluating the empirical power of the four methods - \(\rm HC0\), \(\rm HC1\), \(\rm HC2\) and \(\rm HC3\) - suggested that \(\rm HC3\) is a superior estimate, regardless of the presence (or absence) of heteroskedasticity.
The corrected coefficient estimate variance covariance matrix is used to correct the standard errors (and \(p\)-values) of our coefficients:
Although we have corrected for heteroskedasticity - coefficient significance conclusions remain the same as before. This could mean that the heteroskedasticity is not severe enough to affect our statistical test conclusions.
Correcting for autocorrelation
If our residuals are autocorrelated, or autocorrelated and heteroskedastic - we can use \(\rm HAC\) (Heteroskedasticity-and-Autocorrelation-Consistent standard Errors).
How does our model compare with a simple regression model, where we included only one variable?
Coefficient of determination and information criterion(-s)
Important
When comparing models via \(\rm AIC\), \(\rm BIC\), \(R^2\) and \(R^2_{adj}\) it is important that the dependent variable has the same transformation is all of the models, that are being compared.
Note that the models have the following explanatory variables:
We see that adding additional variables leads to a more accurate model:
While the simple regression model explains around \(25.6\%\) of the variation in \(\log(wage)\), adding more variables increases \(R^2\) to \(0.326\) (the new model explains \(32.6\%\) of the variation in \(\log(wage)\)). Accounting for the additional coefficients, the adjusted \(R\)-squared is \(0.32\).
Dropping insignificant variables and including polynomial terms improves the model even further - the explanatory variables in the final model explain \(34\%\) of the variation in \(\log(wage)\), while the adjusted \(R\)-squared is also larger and is equal to \(0.338\).
In terms of Information Criterions we see that both \(\rm AIC\) and \(\rm BIC\) are lower for our final model, which means that it better describes our data.
Important
Remember - we can compare \(R^2\) and \(AIC/BIC\) for models with the same dependent variable (and its transformation).
Having identified the in-sample adequacy of our model on the modelled (i.e. transformed) data. We now look at the in-sample and out-of-sample accuracy of our model on the original data.
In-sample vs. out-of-sample
While we have examined the accuracy in terms of \(\log(wage)\) models - how well does our final model describe \(wage\) itself? To check this, we can calculate \(R^2_{adj}\) for \(wage\).
In order to do this, we will need to calculate \(R^2_{adj}\) ourselves. To make it easier (and not repeat the same calculation steps every time) we will define a function:
r_sq_adj<-function(actual, fitted, k=1){# k - number of parameters, excluding the intercept RSS<-sum((actual-fitted)^2, na.rm =TRUE)TSS<-sum((actual-mean(actual, na.rm =TRUE))^2, na.rm =TRUE)return(1-(RSS/(length(actual)-k-1))/(TSS/(length(actual)-1)))}
def r_sq_adj(actual, fitted, k =1):# k - number of parameters, excluding the intercept RSS = np.sum((actual - fitted)**2) TSS = np.sum((actual - np.mean(actual))**2)return1- (RSS / (len(actual) - k -1)) / (TSS / (len(actual) -1))
We can verify that this function works, by comparing it with the built-in results:
We see that the in-sample and out-of-sample \(R^2_{adj}\) is much lower for \(wage\) compared to the one for \(\log(wage)\). This is due to the fact that \(\log(wage)\) has a linear, while \(wage\) has an exponential relationship to the explanatory variables.
To compare model prediction accuracy, we will define \(\rm RMSE\) and \(\rm MAPE\) as separate functions:
While \(\rm RMSE\) is more sensitive to the scale of data, both measures show that our final model exhibits similar accuracy for both in-sample and out-of-sample data.
\(\rm MAPE\) expresses accuracy as a percentage of the error - in our dataset a \(\rm MAPE\) value of \(12\%\) for \(\log(wage)\) indicates that, on average, the forecast of \(\log (wage)\) is off by \(12\%\). This translates to \(\approx 38\%\) forecast inaccuracy for \(wage\).
Furthermore, we see that the in-sample and out-of-sample values are close (both \(\rm RMSE\) and \(\rm MAPE\)) and are a contrast to the adjusted \(R^2_{adj.}\) value. One explanations is that the \(R^2_{adj.}\) value compares the ratio of the errors of our model, against the errors from the process average. In case of non-linearity (or back-transformation of the dependent variable), the \(R^2_{adj.}\) might be a poor choice as a measure of model adequacy.